Premium USA-Made Research Compounds
Browse lab-tested peptides, research liquids, capsules and more.
Oral Peptide Delivery: Bioavailability Barriers, Permeation Enhancer Research & Emerging Laboratory Strategies
Oral delivery of peptide compounds remains one of the most stubbornly difficult problems in pharmaceutical research. Despite decades of investigation, most therapeutic peptides are administered parenterally — injected subcutaneously, intravenously, or intramuscularly — simply because the gastrointestinal (GI) tract is, in many ways, a formidably hostile environment for these molecules. Yet the scientific rationale for solving oral peptide delivery is compelling. Improved bioavailability through the oral route would transform how peptide compounds are studied and ultimately applied. What, exactly, makes this so hard? And what emerging strategies are researchers using to overcome the barriers?
The Bioavailability Problem: Why Less Than 2% Typically Survives
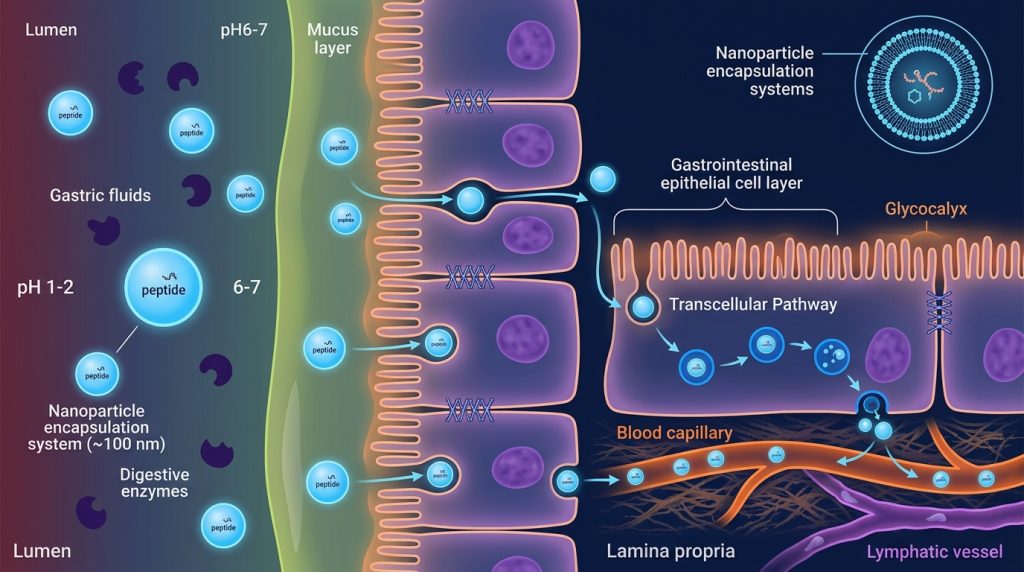
The oral bioavailability of most unmodified peptides is strikingly poor — often below 1–2% of the administered quantity. This is not a single problem but a cascade of overlapping obstacles, each capable of independently eliminating the majority of a peptide molecule before it reaches systemic circulation.
The first barrier is chemical. Gastric pH sits between 1.5 and 2.5, and within this acidic milieu, peptide bonds are susceptible to acid hydrolysis. A peptide that enters the stomach intact may be partially degraded before it even encounters enzymatic activity. Pepsin, the primary gastric endopeptidase, then begins cleaving peptide bonds at aromatic and hydrophobic residues. The attack continues in the duodenum, where pancreatic proteases — trypsin (targeting Lys/Arg), chymotrypsin (aromatic residues), elastase, and carboxypeptidases — constitute a second major enzymatic wave. Brush-border peptidases on the luminal surface of enterocytes represent a final enzymatic checkpoint. By the time a peptide reaches a position where absorption might be possible, the original sequence may be substantially fragmented.
Looking for Premium Research Compounds?
Permeability presents a separate challenge. The gastrointestinal mucosa is designed to be selectively permeable. Molecules with molecular weight exceeding approximately 500 Da are generally poorly absorbed by the paracellular route, and peptides routinely span 1–50 kDa or more. Tight junctions between enterocytes restrict paracellular transport, and the transcellular pathway is complicated further by P-glycoprotein (P-gp) efflux — an ATP-dependent pump that actively expels certain substrates back into the intestinal lumen, effectively reducing net absorption even for compounds that penetrate the apical membrane.
Nanoparticle Encapsulation: Shielding Peptides from the GI Environment
One of the most extensively explored strategies in the literature involves encapsulating peptide compounds within protective nanoparticles or microparticles. The principle is straightforward: if the peptide can be physically isolated from its enzymatic environment until it reaches a more favorable absorption site, degradation losses are reduced.
Poly(lactic-co-glycolic acid) (PLGA) microspheres have received considerable attention for this purpose. PLGA degrades by hydrolysis in a pH- and time-dependent manner, enabling controlled release profiles that can, in theory, be tuned to protect cargo in the stomach while releasing it in the small intestine. Liposomal carriers offer an alternative approach — phospholipid bilayers encapsulating hydrophilic peptides in an aqueous core, with the potential for fusion with intestinal cell membranes. Chitosan nanoparticles are particularly interesting because chitosan is a bioadhesive polysaccharide that adheres to mucosal surfaces and can transiently modulate tight junction permeability, offering a combined protection-and-permeation benefit.
The challenge with nanoparticle platforms is reproducibility and scale. Particle size, surface charge, loading efficiency, and in vivo stability can all vary considerably, and results obtained in rodent models do not always translate predictably.
Permeation Enhancers: Opening the Gate Transiently
A distinct class of delivery strategies focuses not on protecting the peptide from degradation but on improving its ability to cross the intestinal epithelium once intact. Permeation enhancers transiently disrupt tight junctions or alter membrane fluidity to increase paracellular or transcellular flux.
Perhaps the most clinically validated example is SNAC — sodium N-[8-(2-hydroxybenzoyl)amino] caprylate. SNAC is the absorption agent used in the oral formulation of semaglutide (Rybelsus), the first approved oral GLP-1 receptor agonist. Research has shown that SNAC works primarily in the stomach rather than the intestine, creating a locally elevated pH in the gastric mucosa layer that reduces peptic degradation while simultaneously enhancing transcellular permeation at the absorption site. This mechanism — protection plus permeation in a single agent — represents a meaningful conceptual advance.
Cell-penetrating peptides (CPPs) such as TAT (derived from HIV-1 trans-activator protein) and penetratin offer another route. These short, cationic sequences can cross biological membranes via endocytic and non-endocytic mechanisms, and research has investigated their potential as molecular “chaperones” to co-transport larger, membrane-impermeable peptide cargo across the epithelial barrier. The selectivity challenge — CPPs are not tissue-specific — remains an active area of study.
Chemical Modification Strategies: Engineering Protease Resistance
Rather than relying on delivery vehicles to protect a native peptide sequence, chemical modification approaches aim to build resistance directly into the molecule itself. Two strategies have garnered substantial research interest.
D-amino acid substitution replaces one or more L-amino acid residues with their stereoisomeric D-forms. Because most GI proteases are stereospecific — evolved to cleave L-peptide bonds — D-substituted analogs can exhibit dramatically improved resistance to enzymatic degradation while retaining receptor binding affinity. The position and extent of D-substitution must be carefully optimized; indiscriminate substitution can abolish biological activity.
Peptide cyclization — forming a head-to-tail or side-chain-to-side-chain ring structure — constrains the peptide backbone in a conformation that is both more resistant to exopeptidase activity and, in some cases, more complementary to the target binding site. Cyclic peptides are generally less flexible, which reduces the entropic cost of receptor binding and can improve potency. Research into naturally occurring cyclic peptides like the cyclotides (found in certain plant families) has informed synthetic design strategies.
pH-responsive hydrogel systems combine some of these concepts. Hydrogels cross-linked with pH-sensitive bonds contract at low gastric pH, sequestering the encapsulated peptide, then swell in the neutral-to-basic environment of the small intestine to facilitate release — an architecture that exploits the very pH gradient that would otherwise be destructive.
Research Implications and the Epitalon Case Study
Interest in orally deliverable peptides is not merely academic. Search data and research community discussions reveal growing interest in whether specific research peptides — including the tetrapeptide Epitalon (Ala-Glu-Asp-Gly) — might retain meaningful activity when formulated for oral administration. Epitalon’s small size (MW ≈ 390 Da) places it below the conventional permeability threshold, which arguably makes it a more tractable subject for oral delivery research than larger peptides. Whether encapsulation or chemical modification strategies can preserve its bioactivity through the GI transit is a question that ongoing laboratory investigations are beginning to address.
The broader principle here is important: the oral delivery problem is not uniformly difficult across all peptide classes. Molecular weight, charge, hydrophobicity, and sequence composition all influence how a given peptide interacts with the GI environment. Researchers designing oral delivery experiments must characterize their specific compound’s vulnerabilities before selecting a delivery strategy.
Conclusion
Oral peptide delivery sits at the intersection of medicinal chemistry, materials science, and GI physiology — a multidisciplinary challenge that no single strategy has yet fully resolved. Nanoparticle platforms, permeation enhancers, CPP co-administration, D-amino acid substitution, cyclization, and pH-responsive hydrogels each address one or more of the core barriers, but each also carries its own trade-offs and limitations. The success of oral semaglutide with SNAC has demonstrated that meaningful oral peptide bioavailability is achievable — it just requires a deep understanding of the mechanistic landscape. For researchers in this space, the most productive approach is likely a combination strategy: chemical stabilization of the sequence, combined with a targeted delivery vehicle optimized for the specific peptide’s physicochemical properties. The field is moving fast, and the solutions emerging from current research may define how peptide science is conducted for the next decade.
For Research Purposes Only: The information presented in this article is intended solely for scientific research and educational purposes. These compounds are not approved for human use and should only be handled by qualified researchers in appropriate laboratory settings in compliance with all applicable regulations.
Continue Your Research
Explore our complete catalog of premium research compounds.